What is Stargardt disease?
Stargardt disease is the most common form of inherited macular degeneration. It differs from other inherited retinal diseases like retinitis pigmentosa, as it primarily affects the centre of the retina, in a region known as the macula. This means that it affects central vision (reading, face recognition etc), and generally does not affect side vision as much.
Stargardt disease is usually diagnosed in individuals under the age of 20. The progression of visual loss varies between individuals, but peripheral vision is usually preserved. This means that patients rarely have issues with independent mobility.
Fundus flavimaculatus is similar to Stargardt disease but has a later onset and slower progression resulting in a milder condition. Both Stargardt disease and fundus flavimaculatus are linked with mutations in the ABCA4 gene, and so represent variations along a spectrum of disease caused by this gene.
How is Stargardt disease identified?
The presence of Stargardt disease is often identified through the presence of “piscine” or fish-shaped flecks in the retina.
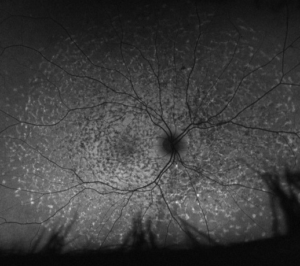
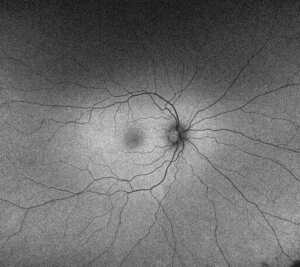
Below are images of a retina with Stargardt disease (below left) compared to a healthy retina (below right).
The retina with Stargardt disease shows white, fish-shaped flecks at the central macular region. Images taken on a wide-view fundus autofluorescence camera (Optos), which shows areas of unhealthy and dead retinal cells more clearly.
Source: Images are courtesy of the VENTURE study.
What are the symptoms of Stargardt disease?
As Stargardt disease affects the macular, symptoms are in the central vision. Common initial symptoms are difficulty reading or recognising faces. Blind spots can occur, and these may increase in size over time. The condition can be slowly degenerative and progressive, but it is very uncommon for someone to become completely blind. Both eyes are usually affected in a similar manner, and colour vision may be affected in the later stages of disease.
The rate of progression and degree of visual loss can vary from person to person, even among affected members of the same family. It is, therefore, very difficult to predict what an individual’s vision will be like at a specific time in the future.
However, Stargardt patients typically experience a rapid decline in central vision between adolescence and young adulthood. The age of onset varies widely with some individuals experiencing delayed vision loss between between the 4th and 7th decades of life. 1
What is the cause of Stargardt disease and how is it inherited?
The prevalence of Stargardt disease worldwide is estimated to be one in 8,000 to one in 10,000. 2 In Australia, this equates to approximately 2,650 to 3,300 affected individuals. It is one of the most common IRDs that leads to blindness, accounting for 12% of all IRD-related blindness. 3
Autosomal recessive inheritance of the ABCA4 gene
The majority of people with Stargardt disease have an autosomal recessive form of disease, involving mutations in the ABCA4 gene. 4
Autosomal recessive inheritance means each parent is a carrier, but is not affected by the disease themselves. Each of their children will have a 25% chance of being affected, with males and females having an equal chance of being affected. The children have a 50% chance of being a carrier (having one healthy and one mutated copy of the gene).
The ABCA4 gene provides instructions to make the ABCA4 protein. If there is a faulty ABCA4 gene, its leads to the build up of a toxic waste product known as A2E in the retinal pigment epithelium (RPE), and can lead to macular degeneration and progressive loss of vision.
Autosomal dominant inheritance of the ELOVL4 gene
A very rare form of Stargardt disease may be caused by mutations in the ELOVL4 gene, which follows an autosomal dominant form of inheritance. In autosomal dominant inherited retinal disease one parent is affected, and each pregnancy has a 50% chance that the child will be affected. Males and females are equally affected. A person only needs one copy of the mutated gene to develop the IRD.
Mutation in the ELOVL4 gene can make dysfunctional ELOVL4 protein clumps that can interfere with photoreceptor cell functions leading to cell death. If a family member is diagnosed with Stargardt disease, it is strongly advised that other members of the family also have an eye exam by an ophthalmologist.
What treatments are available?
At present, there are no effective treatments.
There is research to suggest that UV sunlight can increase the toxicity of the waste products accumulating in the retina 5 6, therefore, it is recommended that people with Stargardt disease wear UV screening sunglasses when out in direct sunlight.
Recent evidence also suggests that taking extra vitamin A, such as vitamin supplements, can be damaging in people with Stargardt disease and should be avoided. 7
A number of emerging treatments are being developed that may assist people with Stargardt disease in the future.