Retinitis pigmentosa (RP) refers to a group of inherited retinal diseases (IRDs) that affect the photoreceptor (light sensing) cells in the retina. People with RP experience a gradual decline in their vision because the two types of photoreceptor cells – rod and cone cells – die over time. RP is often also known as rod-cone dystrophy, reflecting the order of cell death (rods first, followed by cones).
Rod cells are present throughout the retina, except for the very centre, and they help with night vision. Cone cells are also present throughout the retina but are concentrated in the central region of the retina (the macula). They are useful for central (reading) vision and for colour vision. In RP, the rod cells, and eventually the cone cells stop working, which can cause central vision loss and blindness.

How is RP identified?
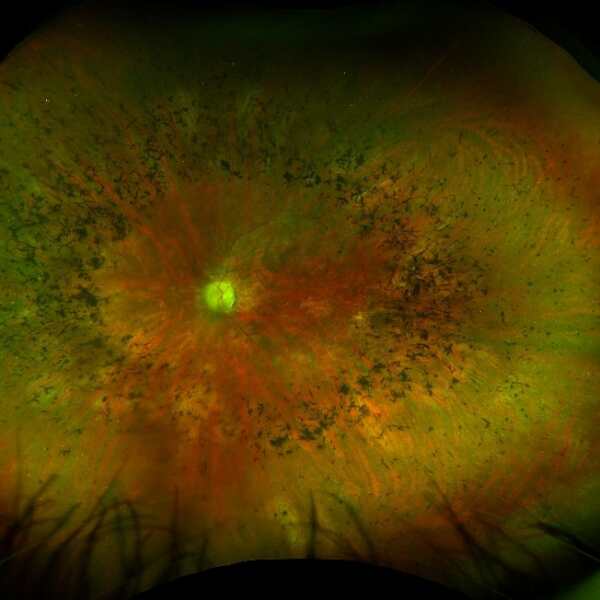
RP is characterised clinically by black splotches of pigment in the outer regions of the retina (called bone spicule pigmentation), thinning of the blood vessels on the retina and a pale appearance of the optic nerve. 1
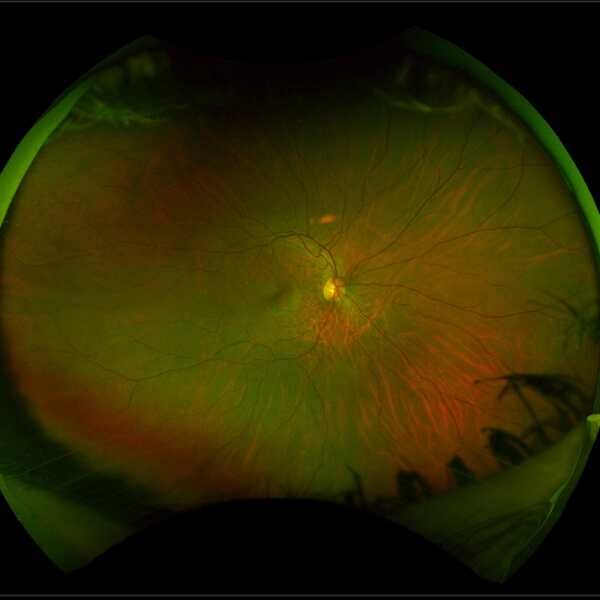
The image below left shows the appearance of a retina with RP compared to the image of a healthy retina below right. The retina with RP shows peripheral bone spicule pigmentation, retinal blood vessel thinning and a pale optic nerve.
Source: Images have been taken on a wide-view fundus camera (Optos) and are courtesy of the VENTURE study.
What are the symptoms of RP?
As the initial damage in RP occurs in the rod photoreceptor cells, the first symptoms are typically difficulty seeing in dim light, including transitioning from light to dark and vice versa. There can be a very variable range in the age of onset of RP. Some people are diagnosed in childhood, while others are not affected until they are adults.
The condition is slowly degenerative, but the rate of progression and degree of visual loss can vary from person to person, and even among affected members of the same family. It is therefore very difficult to predict what an individual’s vision will be like at a specific time in the future. Both eyes are usually affected in a similar way.
As RP progresses, the peripheral vision loss moves slowly inwards, giving people the experience of “tunnel vision”. In some people, the vision loss can eventually cause loss of the central vision too.
What is the cause of RP and how is it inherited?
- In Australia, it is estimated that one in 3,000 people has RP, which equates to approximately 8,900 individuals. 2
- RP is the most common form of IRD. 3
- Over 50 different genes have been identified that can cause RP.
- A number of these were discovered in Australian individuals by the team at the Australian Inherited Retinal Disease Register (AIRDR) based in Perth, Western Australia, which has been given funding grants by Retina Australia over many years.
- It is likely that researchers will discover more genes responsible for RP in the future.
Inheritance patterns of RP
There are various inheritance patterns for RP, including autosomal dominant (30-40%), autosomal recessive (50-60%) and X-linked (5-15%). 4
- In autosomal dominant inherited retinal disease one parent is affected, and each pregnancy has a 50% chance that the child will be affected. Males and females are equally affected. A person only needs one copy of the mutated gene to develop the IRD.
- Autosomal recessive inheritance means each parent is a carrier but is not affected by the disease themselves. Each of their children will have a 25% chance of being affected, with males and females having an equal chance of being affected. The children have a 50% chance of being a carrier (having one healthy and one mutated copy of the gene).
- X-linked conditions are caused by gene mutations on the X-chromosome. In these conditions, an affected male and an unaffected female will have unaffected sons and carrier daughters. In another example, a carrier female with an unaffected male will produce children with a 50% chance that each son will have the inherited retinal disease. There is also a 50% probability that each daughter will be a carrier.
The autosomal dominant form of disease tends to follow a milder course with maintenance of preserved vision well into late middle age. The X-linked form is the most severe and central vision may be lost by the third decade.
RP is a genetic disease, but cases with no family history also commonly occur. If a family member is diagnosed with RP, it is strongly advised that other members of the family also have an eye exam by an eye doctor (ophthalmologist) who is specially trained to detect retinal diseases. Approximately half of RP patients will not have any family history of the condition.
RP is also a symptom of the rare metabolic disorder Refsum disease. This disease, occurring in 1 in 1,000,000 people, can be fatal if not treated, and should be ruled out for all people with RP. 5 More information regarding Refsum disease can be found here.
What treatments are available?
There is currently only one treatment available for a subtype of RP called RPE65-associated retinal degeneration. Luxturna® (voretigene neparvovec-rzyl) is a gene therapy that replaces the RPE65 gene in the eye, thus slowing or halting the progression of the retinal damage. 6
- It has been approved for clinical use by the US Food and Drug Administration (FDA, USA), National Institute for Health Care and Excellence (NICE, UK) and the Therapeutic Goods Administration (TGA) in Australia.
- The RPE65 gene is involved in the production of a protein essential for the functioning of the retina. Luxturna® delivers a functional copy of the RPE65 gene to the retina, restoring the production of the protein and improving vision. 7
- The treatment involves a one-time injection of the therapy into the affected eye, with the benefits expected to last for life.
It is important to note that Luxturna® is only effective for RP caused by RPE65 mutations and is not suitable for other genetic causes of RP. Whilst the outcomes of Luxturna® treatment have been incredibly exciting, the treatment is suitable for less than 2% of people with RP.
Also, as with any medical treatment, there are potential risks and side effects associated with Luxturna®, and it should only be administered by qualified healthcare professionals. Currently, the two locations in Australia where Luxturna® treatment is available are the Sydney Eye Hospital (team led by Professor John Grigg) and the Royal Victorian Eye and Ear Hospital (team led by Dr Thomas Edwards).
To know if you are eligible for this treatment, an ophthalmologist will complete an eye examination and genetic testing to confirm.
Other emerging treatments for RP are currently under development.